药效学是什么?关于药效学的科普介绍

药物效应动力学(pharmacodynamics)简称药效学,是研究药物对机体的作用及作用机制的学科。其内容包括药物作用与机体引起的生物化学和生理学效应,以及药物作用的机制。
药物作用的基本规律
药物作用和药理效应
药物作用(Drug Action)
是指药物对机体的初始作用。如:去甲肾上腺素与血管平滑肌α受体结合。
药理效应(Pharmacological effect)
是指药物作用引起的机体反应。如:去甲肾上腺素与血管平滑肌α受体结合后所引起的血管收缩、血压升高的现。
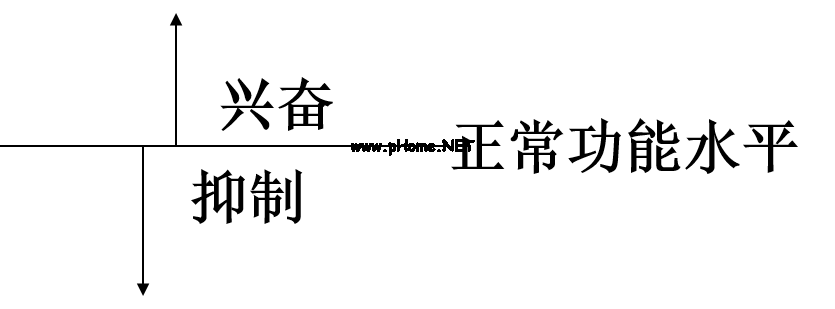
药物作用的基本表现是改变机体原有的功能水平。功能提高称为兴奋(Excitation),功能的降低称为抑制(Inhibition)。肾上腺素升高血压属于兴奋,阿司匹林退热属于抑制。
药物作用具有选择性(selectivity)。
选择性是指药物在治疗剂量下,药物吸收入血后选择性地作用于某一个或几个器官、组织,而对其他的器官、组织不发生作用。这是由于药物对器官、组织的亲和力不同,同时,药物作用的选择性也是药物作用的范围。作用范围窄的药物选择性高,作用范围广的药物选择性低。例如:青霉素能抑制革兰氏阳性菌(Gram-positive Bacteria)的细胞壁合成,其对杀灭革兰氏阳性菌的作用具有高度选择性。
药物作用根据作用范围分为局部作用(Local action)和全身作用(Systemic action)。
局部作用是指药物在用药部位产生的作用。全身作用是指药物自用药部位吸收入血后分布到全身而产生的作用,也称吸收作用。例如:毛果芸香碱滴眼液的缩瞳作用为局部作用,而药物经黏膜吸收后增加唾液分泌的作用则是全身作用。
药物的治疗作用
药物的治疗作用(Therapeutic effect),是指符合用药目的,有利于改变患者的生理、生化功能或者病理过程,使患病的机体恢复正常的药理作用,也称疗效(Symptomatic treatment)。其中,根据治疗作用的效果又可分为对因治疗(Etiological treatment)和对症治疗(Symptomatic treatment)。对因治疗是指用药的目的是为了消除原发致病因子,彻底治愈疾病,或称治本 。对症治疗是指用药的目的是为了改善症状,或称治标。
药物的不良反应
药物在治疗疾病的同时,也会产生不利于机体的反应(Untoward Reaction or AdverseReaction),包括副作用(Side Effect )、毒性反应(Toxic Reaction)、变态反应(Allergy Reaction)、继发性反应(Secondary Reaction)后遗效应(Residual Effect )、致畸作用(Teratogenesis) 等。
副作用
副作用(Side effect)是药物在治疗剂量下出现的与治疗目的无关的作用。副作用对机体的影响较轻,是可逆的,停药后会消失,采取一定的措施可以减轻或者避免。例如:麻黄碱防治支气管哮喘会同时引起中枢兴奋作用从而使患者失眠。
毒性反应
毒性反应(toxic effect)是由于药物剂量过大或者用药时间过长所引起的严重不良反应。毒性反应是药物对机体器官、组织产生的功能性或器质性的损害,一般比较严重,甚至会危及患者生命。短期内大量用药引起的药物毒性反应称为急性毒性(acute toxicity);在长期用药中由于药物在体内蓄积而逐渐发生的毒性反应称为慢性毒性(chronic toxicity)。如:氯霉素能抑制骨髓造血功能,卡那霉素会对肾有损害。
变态反应
变态反应(allergic reaction)是药物引起的免疫反应。反应的性质与药物剂量以及原有效应无关,药物本身、药物辅料、药物代谢物等等都可以是致敏原。机体中抗体的产生需要7-10天的致敏过程,再次与抗原接触时会导致变态反应,变态反应又称为过敏反应(anaphylactic reaction)。变态反应在临床表现的个体差异与反应严重差异程度较大;轻者可出现皮疹、发热等症状,重者则出现哮喘,造血系统以及肝肾功能的损害甚至休克等。例如:患者在使用青霉素之前必须做皮肤过敏性测试。
后遗效应
后遗效应(residual effect)是停药后血药浓度降到阈值以下时所残存的药理效应。例如:夜间使用巴比妥类催眠药,次日清晨还有短暂性的乏力、嗜睡反应。
停药反应
停药反应(withdrawal reaction)是长期应用某种药物之后,突然停药后发生原有疾病加重的现象,包括反跳现象和停药症状。反跳现象(rebound phenomenon)是在突然停药后原有疾病加重的现象,例如:长期服用治疗高血压的药物在突然停药后可使去甲肾上腺素释放过多,出现短时交感神经亢奋,血压突然增高;除此之外产生了原有疾病所没有的症状,如:肌肉疼痛、肌肉强直、关节痛、情绪低落等,称为停药症状(withdrawal symptom)。
特异性反应
特异性反应(idiosyncratic reaction),不同与变态反应,是少数患者由于遗传因素所导致地对某些药物的反应性特别强或者出现于正常人不同性质的反应现象。
继发反应
继发反应(secondary reaction)是因药物治疗作用引起的不良后果。例如:长期使用广谱抗生素肠内的菌群环境会被破坏,耐药菌株会大量繁殖引起再度感染。
耐受性
耐受性(tolerance)是连续用药后出现的机体对药物的反应程度降低的现象。若在很短时间内产生则称为急性耐受性(tachyphylaxis),停药后可恢复。若长期用药后产生则称为慢性耐受性(bradyphylaxis)。病原体和肿瘤细胞在长期用药后对药物产生的耐受性称为耐药性或抗药性(resistance)。
依赖性
依赖性(dependence)是药物长期应用后与集体互相作用所造成的一种状态,表现出强迫要求连续或定期使用某药物的行为或者其他反应。依赖性又分为生理依赖性(physical dependence)和精神依赖性(psychological dependence)。生理依赖性在停药后将引发机体的生理功能紊乱,称为戒断综合征(withdrawal syndrome);精神依赖性是指患者在用药后产生的愉悦/渴求感,并在精神上不能自制地周期性或连续性地滥用药物。
药物的量效关系和构效关系
药物的量效关系
药物引起的药理效应依赖于药物剂量(浓度)。在一定范围内,药物剂量(浓度)增加或者减少时,药理效应随之增强或减弱。药物这种剂量(浓度)与药理效应之间的关系被称为剂量-效应关系,简称量效关系(Dose-effect relationship)。
量效关系一般用量效曲线(Dose-effect curve)表示。量效曲线以药理效应的强度为纵坐标,药物剂量(浓度)为横坐标。
量反应(Graded response)与质反应(Qualitative response)
量反应,一般用具体数量(百分率)来表示药理效应强弱。
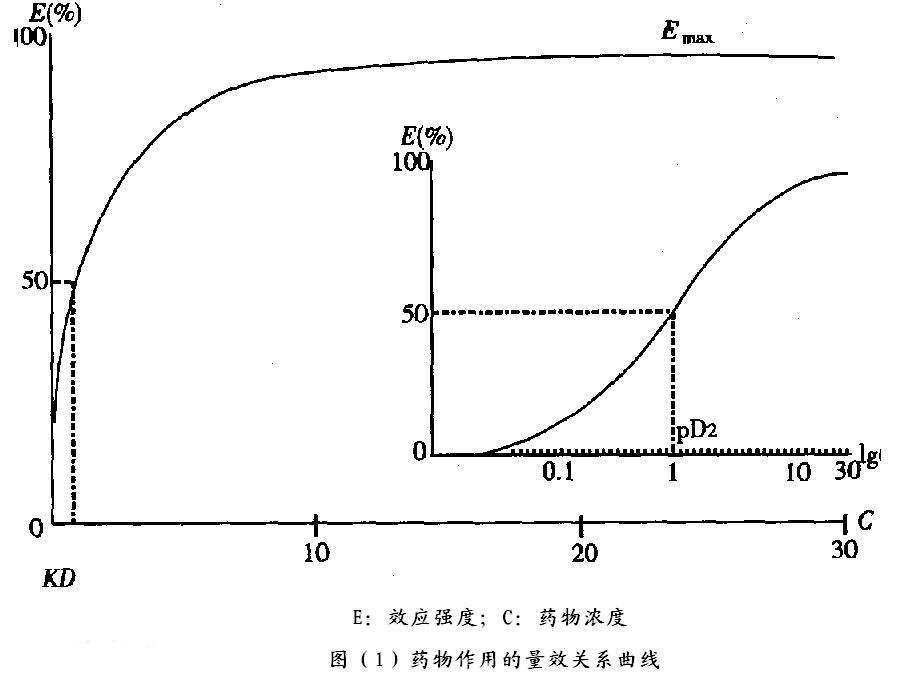
质反应,只以阳性和阴性(有效或无效)来表示药理效应强弱。

量反应的量效曲线
以药物剂量为横坐标,量反应效应为纵坐标所制成的曲线图像称为量反应量效曲线。该曲线一般为不对称曲线,若将横坐标用对数剂量表示,则曲线会呈“S”形。
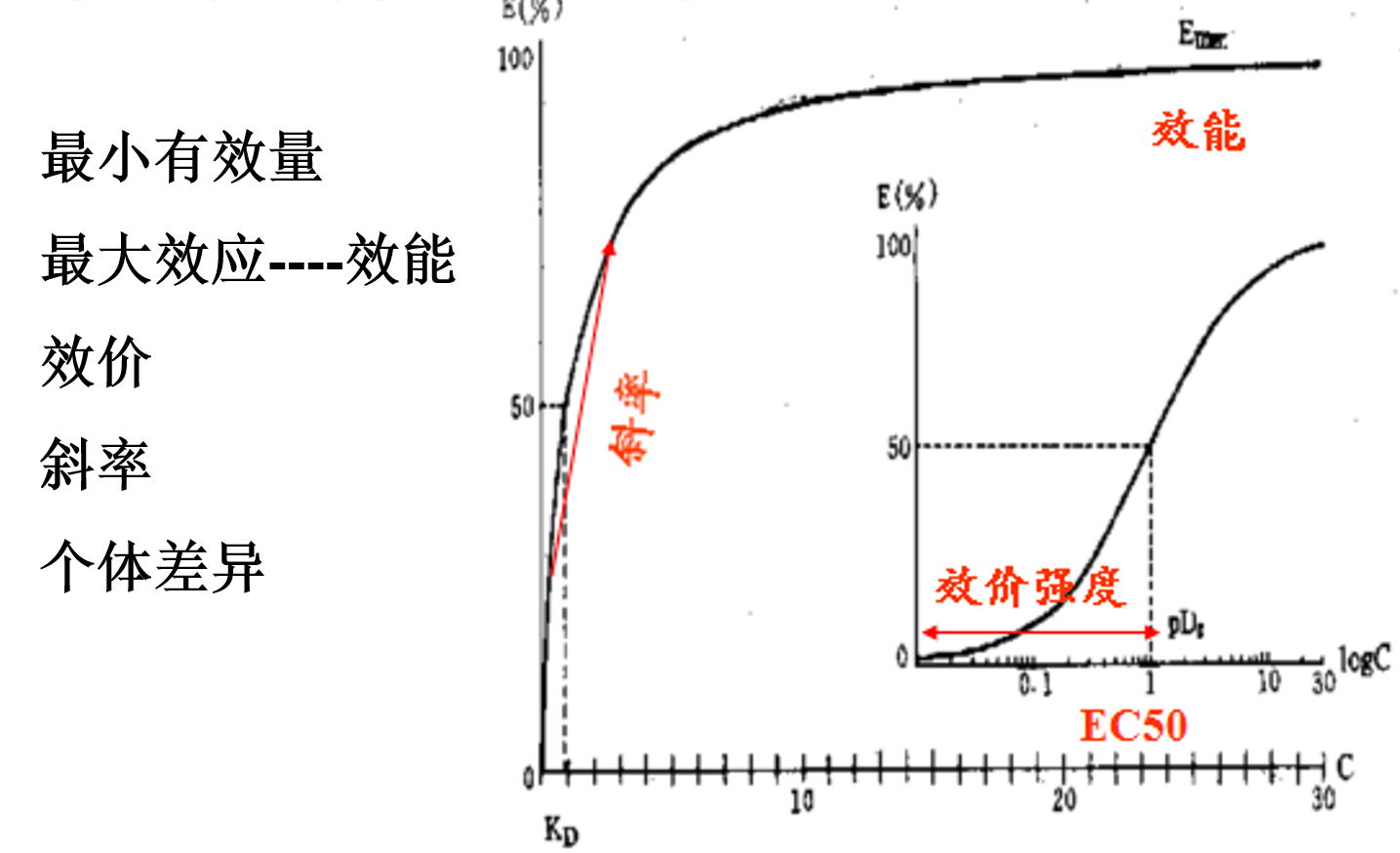
最小有效量(Minimal effective dose,Emin)
最小有效浓度,也称阈剂量或阈浓度。是指引起药理效应的最小剂量或者最低浓度。
效能(Maximum effect,Emax)
是指药物所能产生的最大效应。随着药物计量(或血药浓度)增加,效应浓度相应增强,当效应达到一定程度后,再增加剂量(或血药浓度)效应不会再继续增强,这一药理效应的极限成为效能。
效价强度(Potency)
是指能引起等效反映的剂量,其值小则效价强度越大。药效性质相同的两个药物的效价强度进行比较称为效价比。如10mg吗啡的镇痛作用强于哌替啶100mg的镇痛作用强度相当,及吗啡的效价强度为哌替啶的10倍。
斜率(Slop)
量效曲线在效应量16%~84%时大致呈直线,该段直线与横坐标夹角的正切值称斜率。斜率大的药物当药量微小变化时,即可引起效应的明显变化。因此,斜率的大小是确定临床用药剂量范围的依据之一。
个体差异(Individual variability)
是指药理效应在不同个体间存在差异的现象。量效曲线上任何一点都可以有4个方向的变异,即同剂量可引起不同效应。而相同效用可由不同剂量引起。
质反应量效曲线
以药物剂量(浓度)为横坐标,阳性反应频数为纵坐标作图得到质反应的量效曲线。若纵坐标为累计阳性反应百分率,则可得“S”形曲线;若以药物浓度(剂量)的区段出现阳性反应的频率作图,则可得正态分布曲线。
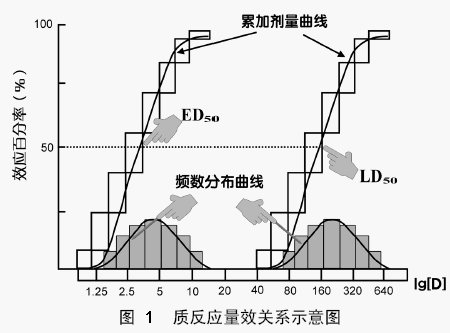
半数有效量(Median effective dose,ED50)
使群体中半数个体出现某一效应的剂量 。
半数中毒量(Median toxic dose,TD50)
使群体中有半数个体出现中毒的剂量 。
半数致死量(Median lethal dose,LD50)。
使群体中有半数个体出现死亡的剂量。
治疗指数(therapeutic index,TI)
是表示药物安全性的指标,TI=LD50/ED50,此数值越大,表示有效剂量与中毒剂量(或致死剂量)间距离越大,越安全。化疗药的治疗指数又称化疗指数(chemotherapeutic index)。
安全指数(safety index,SI)和安全界限(safety margin)。
安全指数=LD5/ED95;
安全界限=(LD1-ED99)/ED99×100%。
临床有时也用药物的安全范围(margin of safety)表示药物的安全性,药物安全范围是指最小有效量和中毒量之间的距离。为了保证临床用药安全,用药时必须一并考虑其治疗指数及安全范围的大小。
药物的构效关系
药物的构效关系(Structure activity relationship, SAR)是指药物的结构与药理效应之间的关系。
化学结构相近的药物常能与同一受体或酶结合,引起相似或相反的作用。
化学结构相同,光学异构体不同,则药理作用不同。
药物化学结构相似,但取代基不同,可影响药物的理化性质和体内过程。
药物作用的机制
改变细胞周围的理化性质
即改变渗透压、影响pH值、络合作用等。如:抗酸药中和胃酸,治疗溃疡病甘露醇在肾小管内提升渗透压而利尿。
补充机体所缺乏的各种物质
除此之外,部分药物还会参与或干扰细胞的代谢。如:铁盐补血,胰岛素治疗糖尿病等,抗代谢药5-氟尿嘧啶与尿嘧啶的结构相似,掺入DNA或RNA中干扰蛋白质合成,而发挥抗癌作用。
对神经递质介质或激素的影响
如:影响递质的合成,摄取,释放,灭活。
作用于特定的靶位
1、药物与受体的相互作用
2、影响离子通道
3、与酶的相互作用
4、影响载体分子
药物与受体
受体与配体
受体(Receptor)是细胞在进化过程中形成的细胞蛋白组分,能识别周围环境中某种微量化合物并与其结合,通过中介的信息传导与放大系统,触发生理反应或药理效应。 能与受体特异结合的物质称为配体(Ligand)。
配体与受体特殊部位结合仅占受体的一小部分,称为特异性结合(Binding site)。
受体的特点
受体具有饱和性(Saturability)
即受体数量是有限的,能结合的配体量也是有限的。
特异性(Specificity)
一种特定的受体只能与特定的配体结合,产生特定的生理效应。因为受体对配体有高度识别能力,对配体的化学结构、立体结构具有很高的专一性。
可逆性(Reversibility)
受体与配体所形成的复合物可以解离,也可被另一种特异性配体所置换。多数通过离子键、氢键、范德华力,是可逆的。
高灵敏性(Sensitivity)
受体能识别周围环境中微量的配体。很低浓度的配体就能与受体结合,产生显著的效应。
多样性(Multiple-variation)
同一受体可广泛分布于不同组织、同一组织的不同区域,受体密度不同。
竞争性(Competition)
结构相似的不同配体可以与统一受体发生竞争性结合。
受体类型
1、G-蛋白耦联受体(鸟苷酸结合调节蛋白)
是通过G蛋白连接细胞内效应系统的膜受体。如M-乙酰胆碱受体、肾上腺素受体等。
2、门控离子通道型受体(离子通道型受体)
是直接连接有离子通道的膜受体,存在快反应细胞膜上,由数个亚基组成,起着快速的神经传导作用。
3、具酪氨酸激酶活性的受体
是结合细胞内蛋白激酶,一般为酪氨酸激酶的膜受体。
4、细胞内受体
调节基因表达的膜受体。

受体的调节
受体上调(Up-regulation)
受体周围生物活性物质浓度低或长期使用拮抗剂,可使受体数量增加(向上调节)、亲和力升高、效应力增强。如长期使用普萘洛尔后突然停药,可出现心率加快,血压升高,心绞痛症状加重现象。因普萘洛尔是β受体阻断药,因长期使用此药,由于受体的向上调节机制,使受体数量增加,当突然停药,可增加机体对肾上腺素、去甲肾上腺素和多巴胺等内源性β受体激动剂的敏感性,故可出现上述反跳现象。
受体下调(Down-regulation)
受体周围生物活性物质浓度高或长期使用激动剂,可使受体数量减少(向下调节)、亲和力下降、效应力降低。如长时间使用α受体激动药可乐定降血压时,当停止使用可乐定,可产生高血压的危险,其中的原因可能是药物下调了α肾上激素受体。
药物与受体相互作用学说
受体理论是药效学的基本理论之一。受体理论从分子水平上阐述机体生理病理过程,药物作用及其机制,药物分子结构与其效应之间的关系。受体假说主要有以下几种。
占领学说(Occupation theory)
1926年clark提出药物与受体之间存在亲和力,1937年,gaddum提出受体占领学说。该学说认为药物必须占领受体才能发挥作用,药物效应与被占领的受体数量成正比,受体全部被占领时,出现最大效应。他认为:药物与受体的相互作用是可逆的;药物效应与被占领受体的数量成正比,当全部受体被占领时就会产生最大效应;药物浓度与效应关系服从质量作用定律;药物效应取决于药物-受体之间的亲和力和药物的内在活性。
故根据药物的内在活性和亲和力又可分为:
完全激动药
为既具有亲和力又具有内在活性的药物,它们能与受体结合并激动受体而产生效应(α=1)。
拮抗药
为只有较强的亲和力而无内在活性的药物(α=0)。
部分激动药
有较强的亲和力但内在活性不强(1>a>0),也称部分拮抗药。
线性关系
Clark发现,许多药物的作用符合这样一种观点,即所观察到的药物效应(E)与被占领的受体的比例呈线性关系,当全部受体(RT)被占领时,药物达其最大效应(E max)。这就是说,被药物占领的受体的比率[DR]/[RT]等于药物所产生的效应与可能产生的最大效应的比率E/Emax,[DR]是药物一受体复合物的浓度。根据激动剂的解离常数KD。
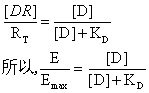
上式预示,以实际反应所占最大反应的分数(分数反应)对激动剂的浓度作图,得一条从原点起始的并逐渐靠近Emax的双曲线;以分数反应对激动剂浓度的对数作图,则得一条对称的S形曲线。当激动剂浓度为使所得反应等于最大反应的一半所需的浓度时,[D] = KD。因此,如果公式[DR]/[RT]=E/E max中所包含的几种假设是正确的,那么,激动剂—受体相互作用的解离常数值可以从E/Emax对[D]或E/Emax对对数[D]作图中读取。也就是说,激动剂与其受体位点相互作用的解离常数(KD),可由产生1/2最大反应的激动剂浓度([Dmax/2])给出。上述一般处理方法的难题是,有些药物无论浓度多大也不能产生最大反应。因而提出“部分激动剂”和“内在活性”的概念。
(2)非线性关系
Stephenson注意到,许多研究结果不符合“药物效应与其所占领受体数目的比例呈线性关系”这一理论。他提出,某些激动剂只占据一小部分受体,就能产生最大效应。未被占领的受体称为“备用受体”(spare receptors)。Stephenson以效能(efficacy)一词表示激动剂占领受体时产生反应的能力。他认为,一个激动剂的活性为其效能与亲和力的乘积,拮抗剂的效能等于零。
速率学说(Rate theory)
1961年,paton提出速率学说(rate theory)。认为药物作用主要取决于药物与受体结合及分离速率,而与药物占领受体量无关。药物作用的效应与其占有受体的速率成正比,即药物分子与受体结合位点相互碰撞时,产生一定量的刺激,并传递至效应器的结果。
二态模型学说(Two model theory)
karlin和changeux根据实验资料,提出了药物一受体相互作用的两态学说,即受体构型存在两种状态,活化态和失活状态,两者可以相互转化,处于动态平衡。
诱导契合学说(The Induced Fit Model)
此外,有新的概念引入认为药物与受体的相互作用主要是依靠以下两种方法:
1、相互作用力契合。
例如:肾上腺素能激动α-和β-肾上腺素受体,而叔丁基肾上腺素仅能激动β-肾上腺素受体。
2、空间形状、诱导契合。
在药物-受体相互作用时,具有柔性的受体结合部位受药物的诱导而发生构像的变化,产生可逆的、互补的契合。
后者则是诱导契合学说的概念。受体学说是建立在早期用来阐明配体-受体结合机制的原理上的,是Fisher于1894年提出的“锁钥模型",配体进入受体的方式类似于锁和钥匙,此时视受体和配体均为刚性结构,即在配体和受体进行分子对接的过程中,空间构象不发生变化。由于“锁钥”原理的局限性,1958年D.E.Koshland提出了“诱导契合"学说,其核心内容为蛋白的活性位点通过与配体的相互作用而发生变化,即蛋白与底物契合并发生结构变化,该原理说明在分子对接的过程中,配体和受体均被视为柔性结构。事实证明,将配体和受体视为柔性结构得到的对接结果相对而言更为准确。
药物受体相互作研究方法
分子对接
分子对接技术属于计算机辅助药物设计(computer-aided drug design,CADD)的一种主要方法,近年来,在筛选中药药效物质、寻找药物作用于疾病的靶点以及探索中药的作用机制被广泛运用并取得了一定进展。分子对接是研究蛋白质-配体分子间相互作用与识别的有效方法。分子间的相互作用过程中形成的近天然构象是结合自由能极低的构象,快速且准确搜索能量极低的构象对于蛋白质-配体分子对接至关重要。
刚性对接
即在对接计算的过程中,参与对接的分子的构象并不发生变化,只有分子的空间位置和姿态发生变化。由于在刚性对接中,受体和配体的空间形状均被视为固定不变的,所以该方法简化程度较高,并且计算难度和计算量都较小,故适用于处理结构比较大的分子,一般应用于模型建立后的初步分析。
半柔性对接
即在对接计算过程中,受体的构象是刚性的,固定不变,只允许配体的构象在一定范围内变化,如固定某些非关键部位的键角和键长等。因为该对接方法兼顾了整个对接计算过程中的计算量和模型的预测能力,因而广泛应用于小分子和大分子(酶或核酸)之间的对接。在药物的研发和设计方面,该方法已得到了广泛的应用,通过固定受体蛋白的构象,并不断调整药物分子的化学结构,使之相契合,从而得到药物分子的初步设计方案。
柔性对接
即在对接计算过程中,允许配体和受体的构象发生自由的变化,由于变量随着体系的原子数呈几何级数增长,因此柔性对接方法对计算机软件、硬件系统要求都比较高,还要求高精度的分子构象,因此其计算量非常大,耗时较多,得到的对接精度也比较高,适用于精确考察分子间的识别情况。
网络药理学研究
网络药理学由英国科学家Hopkins在2007年率先提出, 他的理念为将药理学与网络分析结合,即形成概念—网络药理学 (Network pharmacology) 。通过系统的观察药物对疾病网络的干预与影响, 进而预测药物的治病作用机制, 评估药物的有效性等,分析、模拟预测药物的药理作用机制并通过相应的实验来检验、评估药物的治疗效果、毒副反应及理论机制。
例如, 利用代谢组学技术进行药物成分的分析, 进而利用网络药理学对其活性靶点的寻找等;利用高通量技术, 观察药物对模型的作用或模型对药物的作用, 在生物网络信息学的基础上建立药物—成分—靶点—疾病网络,从而进一步研究药物可能的作用机制。
本文转载于:搜狗科学,baike.sogou.com/kexue/d34555520559894796.htm,本内容使用CC-BY-SA 3.0授权,用户转载请注明出处